
Dent’s Disease: Symptoms, Genetics, and Treatments
[ad_1]
Dent’s Disease
- Alessander Leyendecker Junior
Clinical Aspects
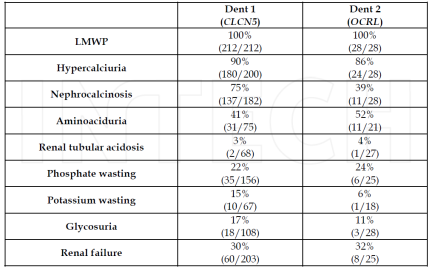
Dent’s disease is a recently characterized syndrome caused by a genetic mutation that leads to a renal tubular disorder and is characterized by manifestations of proximal tubule dysfunction. The proximal tubule is responsable for the reabsorption of sodium, potassium, calcium, glucose and low-molecular-weight proteins (like retinol binding protein and α1 microglobulin and β2 microglobulin) and for the secretion of ammonia, toxins, drugs and H+. Therefore, a proximal tubule dysfunction like Dent’s disease is characterized by low-molecular-weight proteinuria (excretion of low-molecular-weight proteins in the urine), hypercalcinuria (high concentration of calcium in the urine), nephocalcinosis (deposition of calcium salts in the renal parenchyma), nephrolithiasis (formation of crystalline aggregates that can develop anywhere along the urinary tract) and progressive renal failure. Low-molecular-weight proteinuria represents the most common symptom of Dent’s disease (Devuyst & Thakker, 2010).
Mapping:
A higher disease severity in males and a lack of male to male transmission were identified by an analysis of 5 unrelated British families. The fact that females may show a milder phenotype suggests an X-linked recessive inheritance. The locus position on chromossome Xp11 was identified by linkage analysis of 2 3-generation families with Dent disease by Pook et al. (1993). Twenty X-linked polymorphic markers were used and the Linkage was established between Dent’s disease and the Xp1l 1 loci ARAFI (Z max = 5.42, 0 = 0.000), DXS426 (Z max = 3.61, 0 = 0.000), DXS255 (Z max = 5.48, 6 = 0.000) and DXS988 (Z max = 4.25, 0 = 0.045) to determine a locus order. In addition, DXS255 has presented a microdeletion in the affected members of one family, allowing the localization of Dent’s disease to Xp11.22 (Pook et al., 1993). Furthermore, another proximal renal tubular disorder associated with nephrolithiasis in a North American family and referred to as X-linked recessive nephrolithiasis has also been mapped to Xpl1.22. Suggesting that maybe there is one gene causing both X-linked recessive nephrolithiasis and Dent’s disease.
Molecular Genetics:
A study conducted by Fisher et al (1994) found a microdeletion in one Dent’s disease kindred, allowing the identification of the gene CLCN5 as a candidate. Furthermore, an investigation of 11 kindreds with renal tube disorders indentified 3 nonsense, 4 missense, 2 donor splice site mutations, 1 intragenic deletion and 1 microdeletion in CLCN5 gene (Llyod et al., 1996).

Furthermore, Hoopes et al. (2005) indentified that some patients with Dent’s disease suffer from extra-renal symptoms such as cataract, mild intellectual impairment and hypotonia. Shared mutations in OCRL1 with the oculo-cerebrorenal syndrome of Lowe were identified in these patients. The presence of such extra-renal symptoms with mutations relating to Lowe syndrome is known as Dent disease 2.
A research conducted by Bökenkamp et al. (2009) showed that low-molecular-weight proteinuria was a constant feature in all both Dent disease 1, Dent disease 2 and oculo-cerebrorenal syndrome of Lowe. However, there was a wide overlap in most of the other symptoms of the proximal tubulopathy. Nephrocalcinosis was observed more frequently in patients with Dent’s disease 1 than those with Dent’s disease 2 and Lowe syndrome; aminoaciduria, renal tubular acidosis, and renal failure was observed more frequently in patients with Lowe syndrome than patients with Dent-disease 1 and Dent disease disease 2.
Genetics:
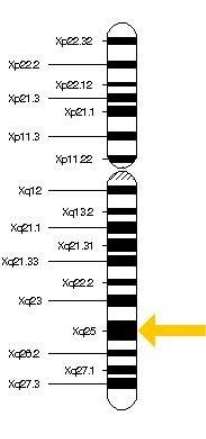
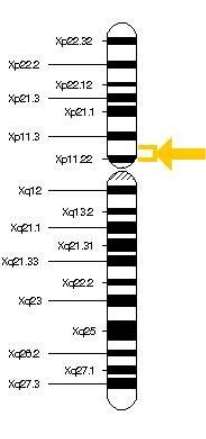
All these findings have demonstrated that both Dent disease 1 and Dent disease 2 are a X-linked recessive inherited condition, caused by mutations in the OCRL1 (Dent disease 2) or CLCN5 (Dent disease 1) genes. These genes are located on chromosome Xp11.22 (Dent disease 1) and Xq25 (Dent disease 2).
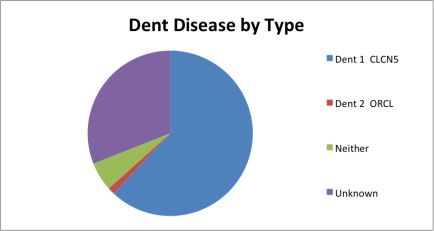
Mutation in CLCN5 are responsible for about 60% of the cases of Dent disease and mutation in OCRL are responsible for about 15% of the cases of Dent disease (Hoopes et al., 2004). So, nearly 25% of the cases of Dent disease do not have a known disease-causing mutation in OCRL or CLCN5, implying another genetic heterogeneity.
Due to the fact that Dent’s disease is a X-linked recessive disorder, the disease is generally found in males only. Females carriers may show a milder phenotype. Furthermore, the disease may be present in early childhood (Devuyst & Thakker, 2010).
To date around 250 families with Dent-1 disease and about 50 patients with Dent-2 disease have been reported (Ludwig, Levtchenko & Bökenkamp, 2014). The diagnosis of Dent’s disease is always difficult due to the wide variability of clinical presentation and, occasionally, lack of family history. Therefore, the disorder is likely underdiagnosed and the prevalence of this disease in the population is still unknown.
More than 200 Dent’s disease 1 patients withCLCN5defects have been described with several different disease-causing mutations all over the gene. The types of mutations includes missense (44%) and nonsense (26%) mutations, small deletions/insertions (15%) and splice defects (11%), with a few hotspots, usually affecting arginine codons. Large insertions/deletions can be detected in around 4%of the patients. However, OCRLmutations in Dent’s disease 2 patients are not uniformly distributed. Missense mutations are usually found in exons 8–15 while nonsense or frameshift mutations very often affect exons (Ludwig, Levtchenko & Bökenkamp, 2014). CLCN5 encodes the electrogenic Clâ»/H+ exchanger ClC-5. OCRL1 encodes a phosphatidylinositol bisphosphate (PIPâ‚‚) 5-phosphatase and mutations are also linked to the Lowe Syndrome. Mutations in any of these genes can culminate in the lack or dysfunction of these important proteins and result in the phenothype of Dent’s disease.
Pathophysiology:
The ability to reabsorb low-molecular-weight proteins and albumin that are ultrafiltered by the glomerulus are characteristics of the epithelial cells covering the proximal tubule portions of the nephron. This ability involves a process called receptor-mediated endocytosis. In the receptor-mediated endocytosis the particle to be endocytosed binds to specific receptor proteins concentrated at certain locations in the plasma membrane. Several types of receptors participate in the receptor-mediated endocytosis. These sites form a small depression in the plasma membrane that is covered by clathrin (a type of fibrous protein). After the binding of a ligand to plasma membrane-spanning receptors, a signal is sent through the membrane, leading to membrane coating, accumulation of the ligands into coated pits, and formation of a membrane invagination. The receptor and its ligand are then opsonized in clathrin-coated vesicles. The clathrin confers stability to the vesicle that are being transported into the cell. In the cytoplasm the vesicle loses its clathrin coverage and individual vesicles fuse to early endosomes. The acidification of endosomes by proton pumps ATP-dependent leads to the dissociation of the protein-receptor complex. This process allows the endosomes to fuse with the lysosomes through late endosomes. This acidification is reached by ATP-mediated transport of cytosolic H+ through the V-ATPase and demand an equal Cl– conductance to preserve electroneutrality. It has been theorized that the H+ gradient can be neutralized by the C1C-5 action. In theory, ClC-5 provides an electrical shunt to neutralize the H+ gradient. Therefore, the vesicular acidification should be impaired by the loss of the endosomal Cl– conductance mediated by ClC-5, resulting in the dysfunction of proximal tubule cells. To confirm this hypothesis, Piwon et al. (2000) created two distinct strains of ClC-5 knock-out (KO) mice which both have symptoms of the main characteristics of Dent’s disease including manifestations of proximal tubule dysfunction like low-molecular-weight proteinuria. A reduced acidification of early endosomes in ClC-5-deficient mice have been demonstrated by in vitro experiments. However, ClC-5 is a 2Cl–/H+ exchanger and not just a Cl– channel, so the importance of this exchange activity for Dent’s disease was still unknown and needed to be assessed. In order to investigate this relevant question, Novarino et al. (2010) created a knock-in (KI) mouse presenting a point mutation in a important glutamate residue which transforms the exchanger into an uncoupled Cl– channel that should help the endosomal acidification. The conventional ClC-5 KO mouse was subsequently compared with the KI mice. Acidification of the renal endosomes from wild-type and KI mice was regular, but greatly impaired in KO mice. Nevertheless, the same symptoms were indentified in both KI and KO mice, despite normal endosomal acidification and patients with Dent’s disease, including low-molecular-weight proteinuria, hypercalciuria and hyperphosphaturia. Moreover, both the KI and KO mouse presented impaired proximal tubule endocytosis, indicating that proximal tubule dysfunction in Dent’s disease may happen in spite of the regular acidification of the endosomes. These findings indicate a function for a diminished endosomal Cl– accumulation in Dent’s disease.
Diagnosis:
The clinical diagnosis of Dent’s disease is grounded on the presence of low-molecular-weight proteinuria, hypercalciuria, and at least one of the following characteristics: kidney stones, hematuria, nephrocalcinosis, renal insufficiency or hypophosphataemia (Hoopes et al., 2004). The clinical diagnosis can also be supported by nephrolithiasis and/or history of X-linked inheritance of renal Fanconi syndrome. The diagnosis can be confirmed by the recognition of mutation in both CLCN5 or OCRL1 by sequence analysis. Sequence analysis of affected males allows the identification of whole-gene deletions, multiexonic and exonic due to the lack of amplification is this case. Confirmation may also require deletion/duplication studies. It is imperative to previously identify the disease-causing mutation in the family when conducting a carrier test for at-risk relatives. The diagnosis of Dent disease 2, related with OCRL1 mutations, is more common than a diagnosis of Lowe syndrome when there are an absence of clinical symptoms and lack of severe intellectual deficit.
Treatment:
The reduction of hypercalciuria, prevention of nephrocalcinosis and kidney stones, and retardate the evolution of chronic kidney disease (nephrolithiasis) are the objectives of the current treatment. Thiazide diuretics in doses higher than 0.4 mg/kg/day have reducted urinary calcium excretion by more than 40% in males with Dent disease (Raja et al., 2002). However, side effects like hypokalemia, volume depletion, and cramping often occur. Similarly, we need to be cautious due to the fact that an increased hypercalciuria can result from the treatment of rickets with vitamin D. Furthermore, recent studies has been shown that a high citrate diet can slow the progression of chronic kidney disease in CLCN5 knockout mice (Cebotaru et al., 2005) and can be efficiently used to treat Dent disease.
Genetic Counseling:
The two types of Dent’s disease are X-linked. Therefore, men are affected more harshly. The father of an affected man will not have the disease or will he be a carrier of the mutation. The probability of transmitting the disease allele in each pregnancy is 50% if the mother of the proband is a carrier. Males who inherit the mutation will be affected and females who inherit the mutation will be carriers and will generally not be strongly affected. Affected men pass the disease-causing mutation to all of their daughters (who become carriers) and none of their sons. It is possible to conduct carrier testing for at-risk female relatives and prenatal testing for pregnancies at increased.
Word count: 1673
References
Blanchard A, Vargas-Poussou R, Peyrard S, Mogenet A, Baudouin V, Boudailliez B, Charbit M, Deschesnes G, Ezzhair N, Loirat C, Macher MA, Niaudet P, Azizi M: Effect of hydrochlorothiazide on urinary calcium excretion in Dent disease: an uncontrolled trial. Am J Kidney Dis 2008, 52:1084-1095.
Bökenkamp A, Böckenhauer D, Cheong HI, Hoppe B, Tasic V, Unwin R, Ludwig M. (2009). Dent-2 disease: A mild variant of Lowe Syndrome. J. Pediatr, 155, pp. 94-99, ISSN 0022-3476
Cebotaru V, Kaul S, Devuyst O, Cai H, Racusen L, Guggino WB, Guggino SE: High citrate diet delays progression of renal insufficiency in the ClC-5 knockout mouse model of Dent’s disease. Kidney Int 2005, 68:642-652.
Devuyst, O., & Thakker, R. V. (2010). Dent’s disease. Orphanet Journal of Rare Diseases.
Fisher, S. E., Black, G. C., Lloyd, S. E., Hatchwell, E., Wrong, O., Thakker, R. V., & Craig, I. W. (1994). Isolation and partial characterization of a chloride channel gene which is expressed in kidney and is a candidate for Dent’s disease (an X-linked hereditary nephrolithiasis). Human molecular genetics, 3(11), 2053-2060.
Hara-Chikuma M, Wang Y, Guggino SE, Guggino WB, Verkman AS: Impaired acidification in early endosomes of ClC-5 deficient proximal tubule. Biochem Biophys Res Commun 2005, 329:941-946.
Hoopes, R. R., Raja, K. M., Koich, A., Hueber, P., Reid, R., Knohl, S. J., & Scheinman, S. J. (2004). Evidence for genetic heterogeneity in Dent’s disease.Kidney international,65(5), 1615-1620.
Hoopes RR Jr, Shrimpton AE, Knohl SJ, Hueber P, Hoppe B, Matyus J, Simckes A, Tasic V, Toenshoff B, Suchy SF, Nussbaum RL, Scheinman SJ. Dent Disease with mutations in OCRL1. Am J Hum Genet. 2005;76:260–267. doi: 10.1086/427887.
Ludwig, M., Levtchenko, E., & Bökenkamp, A. (2014). Clinical utility gene card for: Dent disease (Dent-1 and Dent-2).European Journal of Human Genetics.
Lloyd, S. E., Pearce, S. H., Fisher, S. E., Steinmeyer, K., Schwappach, B., Scheinman, S. J., … & Thakker, R. V. (1996). A common molecular basis for three inherited kidney stone diseases. Nature, 379(6564), 445-449.
Novarino G, Weinert S, Rickheit G, Jentsch TJ: Endosomal chloride-proton exchange rather than chloride conductance is crucial for renal endocytosis. Science 2010, 328:1398-1401.
Pagon, R. A., Adam, M. P., Bird, T. D., Dolan, C. R., Fong, C. T., Stephens, K., & Rossetti, S. (2012). Dent Disease.
Piwon N, Gunther W, Schwake M, Bösl MR, Jentsch TJ: ClC-5 Cl-channel disruption impairs endocytosis in a mouse model for Dent’s disease. Nature 2000, 408:369-373.
Pook, M. A., Wrong, O., Wooding, C., Norden, A. G. W., Feest, T. G., Thakker, R. V. Dent’s disease, a renal Fanconi syndrome with nephrocalcinosis and kidney stones, is associated with a microdeletion involving DXS255 and maps to Xp11.22. Hum. Molec. Genet. 2: 2129-2134, 1993.
Raja KA, Schurman S, D’Mello R G, Blowey D, Goodyer P, Van Why S, Ploutz-Snyder RJ, Asplin J, Scheinman SJ. Responsiveness of hypercalciuria to thiazide in Dent’s disease. J Am Soc Nephrol. 2002;13:2938–44
Wrong.O.M., Norden.A.G.W. and Feest.T.G. (1990) Quart. J. Med. 77, 1086-1087.
The post Dent’s Disease: Symptoms, Genetics, and Treatments appeared first on mynursinghomeworks.
[ad_2]
Source link
